- Database
- Open access
- Published:
AntiJen: a quantitative immunology database integrating functional, thermodynamic, kinetic, biophysical, and cellular data
Immunome Research volume 1, Article number: 4 (2005)
Abstract
AntiJen is a database system focused on the integration of kinetic, thermodynamic, functional, and cellular data within the context of immunology and vaccinology. Compared to its progenitor JenPep, the interface has been completely rewritten and redesigned and now offers a wider variety of search methods, including a nucleotide and a peptide BLAST search. In terms of data archived, AntiJen has a richer and more complete breadth, depth, and scope, and this has seen the database increase to over 31,000 entries. AntiJen provides the most complete and up-to-date dataset of its kind. While AntiJen v2.0 retains a focus on both T cell and B cell epitopes, its greatest novelty is the archiving of continuous quantitative data on a variety of immunological molecular interactions. This includes thermodynamic and kinetic measures of peptide binding to TAP and the Major Histocompatibility Complex (MHC), peptide-MHC complexes binding to T cell receptors, antibodies binding to protein antigens and general immunological protein-protein interactions. The database also contains quantitative specificity data from position-specific peptide libraries and biophysical data, in the form of diffusion co-efficients and cell surface copy numbers, on MHCs and other immunological molecules. The uses of AntiJen include the design of vaccines and diagnostics, such as tetramers, and other laboratory reagents, as well as helping parameterize the bioinformatic or mathematical in silico modeling of the immune system. The database is accessible from the URL: http://www.jenner.ac.uk/antijen.
Introduction
There is a vast, and ever increasing, volume of important information that has accumulated from decades of experimental analysis within immunology. This will only become compounded as high-throughput techniques begin to impinge upon the immunological biosciences. The only efficient way for this information to be properly utilized requires the development of databases that store it and systems that use it. Although the type of data archived may alter from case to case, nonetheless the creation, use, and manipulation of databases containing biologically important information is the most crucial feature of current bioinformatics, both as it supports the genomic and post-genomic revolutions and as a discipline in its own right. There is nothing new in developing databases focusing on immunology: many spotlighting the in-depth sequence analysis of individual immunomacromolecules have existed for some time [1]. Functional or epitope-orientated databases are a more recent development. Examples include the now defunct MHCPEP database [2]http://wehih.wehi.edu.au/mhcpep/, FIMM [3]http://sdmc.krdl.org.sg:8080/fimm, SYFPEITHI [4]http://www.syfpeithi.de, the HIV sequence database [5]http://hiv-web.lanl.gov/, the HLA ligand database [6]http://hlaligand.ouhsc.edu, the EPIMHC database [7]http://bio.dfci.harvard.edu/epimhc/, and the MHCBN database [8]http://www.imtech.res.in/raghava/mhcbn/.
An epitope is any molecular structure that can be recognised by the immune, or other biological, system. Epitopes, or the antigen from which they are derived, can be composed of protein, carbohydrate, lipid, nucleotide, or a combination thereof. It is through recognition of foreign, or non-self, epitopes that the immune system can identify and, hopefully, destroy pathogens. Hitherto, peptide epitopes have been the best studied, and have, traditionally, have been categorized as either T cell or B cell epitopes. T cell epitopes are peptides presented to the cellular arm of the immune system via the MHC-peptide-TCR complex. B cell epitopes represent surface regions of an antigen that are bound by soluble or membrane-bound antibodies. If this region of a protein antigen is comprised of residues distally separated within the primary structure, and brought into local proximity by protein folding, then it is termed a discontinuous or conformational B cell epitope. Linear or continuous B cell epitope residues are sequential in both primary structure and thus as a region on the proteins' surface. Such epitopes are predominantly identified by antigen-specific antibody cross-reactivity with peptides.
There is a need to create a databank for the wider disciplines of immuno-vaccinologists, which can act as a central repository and resource. Our aim is to complement other databanks [2–8] and thus we have developed AntiJen, a computational information resource for immunology and vaccinology that integrates quantitative kinetic, thermodynamic and biophysical data, with functional and cellular information. AntiJen v2.0, a development of our earlier database system JenPep [9, 10], contains functional data on T cell and B cell epitopes. Moreover, the B cell archive is now sub-divided into linear and conformational epitopes. These epitopes form the basis of the humoral immune response and, unlike T cell epitopes, methods of prediction are often inaccurate [11]. A more in-depth B cell epitope archive should aid the development of prediction strategies. Antigen recognition by the Major Histocompatibility Complex (MHC) is vital to T cell activation hence, the inclusion of thermodynamic data on the binding of peptides to MHC molecules and T Cell Receptor (TCR) binding to peptide-MHC (pMHC) complexes. This data is complemented by kinetic data based on the same molecular interactions. Data on antigen processing and presentation is also included in AntiJen. Binding data derived from peptide interactions with the Transporter Associated with Antigen Processing (TAP transporter) are included in the archive. Additionally, quantitative specificity data from position-specific peptide libraries is included. AntiJen also incorporates thermodynamic data on protein-protein interactions, within an immunological context, such as co-receptor and superantigen binding, plus interactions with the MHC. All of these interactions are, potentially, key factors for the successful computational design of vaccines.
AntiJen also contains biophysical data, including diffusion coefficients and cell surface copy numbers, on a variety of immunological molecules. Such data provides insight into the number of target receptors, which is an important, if under explored, component of binding between cells. Indeed, the number of molecules expressed on the membrane can alter depending on disease. The final addition to the databank focuses upon antigen binding to antibodies. One key innovation is a greatly increased compendium of experimental conditions, which, in conjunction with a greatly enhanced search capacity, consolidates our databases as a unique, value-added data source, fostering developments within both in silico immunology and the wider community of immunovaccinology. The database is available from the URL: http://www.jenner.ac.uk/antijen.
Database development
Relative to the database system used for JenPep [9, 10], the interface to AntiJen is entirely new, having been completely rewritten. AntiJen has been designed and implemented using postgreSQL, a system comprising a relational database and database server, and has thus established increased database robustness, creating an improved infrastructure for foreseeable issues of data storage and data growth. Data within AntiJen is structured into twenty-four normalised tables. Each is category specific and holds either statistical or experimental data. Additional tables accommodate the keyword data – which powers our protein-orientated antigen search and allows integration of the BLAST search – and there is also a structural data table to accommodate links to external structural databases. The user interface consists of a series of HTML forms. The search requests from these forms target PERL scripts integrated with SQL which in turn query the database.
Database content
Compared to its progenitor JenPep [9, 10], the data archived in AntiJen v2.0 has grown considerably in depth (additional data types such as experimental conditions), breadth (addition of new data to existing databases), and scope (addition of extra sub-databases containing novel kinds of information). Additions to AntiJen have been derived from exhaustive searching of the primary literature, to give a dataset of > 31,000 entries. AntiJen v2.0 now consists of 11 sub-databases; details of the different databases are given in Table 1. The relative sizes of the databases and the growth from JenPep are summarised in Table 2.
AntiJen contains both generic and dataset-specific data. For each entry, we record the peptide sequence (eg. YTSDYFISY) of the epitope using the standard one-letter code, its length (9 in this case), and, by linking to the sequence database Swiss-Prot http://us.expasy.org/sprot/ or NCBI http://www.ncbi.nih.gov/entrez, the antigen to which the peptide sequence most closely matches (in the case of YTSDYFISY: C-ests-1 (p54), SWISS-PROT code P41156). The description of the antigen is, wherever possible, obtained directly from the literature. AntiJen is also linked to PUBMED. This allows us to record the original citation associated with the data. For example, for YTSDYFISY, we cite: Journal of Immunol 1994 volume 152 pages 3913–3924, PUBMED ID 8144960. For the T cell epitope, MHC ligand, and TCR-pMHC complex categories, we also record, for each peptide, the MHC restriction in terms of the host species, class (class I vs. class II), and, where the data is available, the serotype and allele. For YTSDYFISY, these data would be human, class I, HLA-A1, and A*0101.
Entries within AntiJen are, in turn, linked to external databases, which enables further in-depth cross referencing. As we have said, protein sequence identifiers, which may be the source of an antigenic peptide or immunological co-receptor, link directly to details in the Swiss-Prot database [12] or the NCBI protein database. Journal references can be viewed via a link to the PUBMED database http://www.ncbi.nlm.nih.gov/pubmed, and thus to full literature references, where available. AntiJen also links to structural data, currently derived from the MPID database http://surya.bic.nus.edu.sg/mpid/[13] and the Protein Data Bank http://www.rcsb.org/pdb/[14]. The database aims to provide access to background data where available.
Allele identifiers serve as a link to the IMGT/HLA database http://www.ebi.ac.uk/imgt at the European Bioinformatics Institute [15]. Inherent variability in the way MHC alleles are named within the primary literature prevents us from unambiguously standardizing nomenclature within AntiJen. HLA nomenclature follows that of the HLA Informatics Group http://www.anthonynolan.org.uk/HIG/. An allele is named using a defined pattern. For example, for HLA-A*0101: the HLA-A refers to the HLA locus; the first 01 to the serologically recognized A1 antigen and the final 01 to the individual HLA allele protein sequence. AntiJen stores the antigen classification (i.e. HLA-A1) and, when available, the specific allele. We have often encountered problems with the nonstandard allele reporting. A 4-digit HLA name necessarily implies the two digit serological antigen, a two digit classification clearly does not imply a specific allele.
During database compilation, a sequence search allows us to identify the protein from which an epitope sequence originates. However, because epitopes are generally short, their sequences may be present in several potential antigens: in orthologues, paralogues, or in totally unrelated sequences. As epitopes are processed from whole proteins via a complex proteolytic pathway, one can use the sequence context to infer preferred proteasomal or endoplasmic protease cleavage patterns, but not if its context is defined incorrectly. Moreover, assuming that AntiJen is used subsequently to assign the antigenic status of proteins, wrongly identifying particular proteins as antigens can lead to the percolation of annotation errors [16, 17].
AntiJen is, where possible, a quantitative database archiving continuous measures of binding. This is a fundamental feature of several sub databases, such as the MHC ligand and pMHC-TCR databases. The binding of an immunological macromolecule to a peptide or other biomacromolecule is quantified as are other receptor-ligand interactions:
R+L↔RL
Here R is the receptor (an MHC or TCR), L the ligand (peptide or pMHC), and RL, the receptor-ligand complex (pMHC or pMHC-TCR complex). The rate of the forward reaction is proportional to [L] [R]. The rate of the reverse reaction is proportional to [RL] as no other species are involved in dissociation. At equilibrium, the forward and reverse rates are equal, and so using kon and koff as the respective constants:
k on [R][L] = k off [RL]
Rearranging:
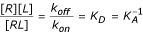
Here KD is the equilibrium dissociation constant, which represents the concentration of ligand that occupies 50% of the equilibrium receptor population, and KA is the equivalent association constant.
Experimentally, the measurement of equilibrium dissociation constants is often addressed using radio-ligand binding assays. Saturation radio-ligand binding assays measure equilibrium binding, at a range of peptide concentrations, to establish affinity (Ka) and receptor number (Bmax). Competitive binding experiments determine binding at a single labelled ligand concentration in the presence of a range of concentrations of unlabelled ligand. AntiJen records a hierarchy of different binding measures in its different sub-databases. Equilibrium constants are the most dependable and sit atop this hierarchy. Next come IC50 values, which can be obtained from a competitive radio-ligand or fluorescence assay. These are the Fmost commonly reported binding measures.
Values obtained from radio-ligand or fluorescence methods may be significantly different. IC50 values for a peptide may vary between experiments depending on the intrinsic affinity and concentration of the standard radiolabelled reference peptide, as well as the intrinsic affinity of the test peptide. IC50 values vary with the equilibrium dissociation constant, at least within a single experiment. In practice, the variation in IC50 is often small enough that values can be compared between experiments. For the peptide discussed above, YTSDYFISY, the radiolabelled IC50 value recorded in AntiJen is 5.3 nM. BL50 values are also obtained from a peptide binding assay and are commonly reported. They are the half maximal binding levels calculated from mean fluorescence intensities of peptides binding to MHCs bound on the surface of RMA-S or T2 cells. Cells, pre-incubated with peptides, are labelled with a fluorescent monoclonal antibody. An overview of the thermodynamic and kinetic binding data within AntiJen is given in Table 3.
AntiJen also now contains experimental conditions, such as temperature and pH. A summary of this data is given in Table 4. The accuracy of data depends greatly upon the experimental method used. The grouping of data with respect to specific experimental techniques allows a more thorough assessment of training sets. Figure 1 shows the distribution of data for each type of analysis with respect to each database. The MHC Kinetics and TAP databases highlight the problems outlined above. The kinetics database contains data determined from over 14 methods while the TAP database is derived from 4 methods, with radiolabelled assays accounting for 80% of the data.

The distribution of experimental methods applied within each database. The number of different experimental methods and the abundance of data relating to the method is shown within the figures. The 'OTHERS' category refers to methods for which there is a relatively small number of entries.
The compilation process has highlighted the considerable inconsistency within the immunological literature regarding the recording of such fundamental data. AntiJen contains, however, a direct, verbatim transcription of data from the primary literature. As such, we do not attempt, as a matter of policy, the comprehensive and retrospective correction of potential errors. To undertake such correction would only compound any errors, introducing the kind of percolating inconsistencies so much a feature of other database systems [16, 17]. Further inaccuracies may stem from our logistic inability to verify data independently, therefore we must trust those values reported in the literature.
Subsidiary Databases in AntiJen
The AntiJen database contains a number of sub-databases. Each of these contains data on different aspects of the biological function and/or biophysical properties of different classes of immunomacromolecule. We describe the nature and content of each sub database below.
B Cell Epitopes
Epitopes are the principal chemical moieties recognized by the immune system. Although the importance of non-peptide epitopes, such as carbohydrates and lipids, is now increasingly well understood, peptidic B cell and T cell epitopes remain the principal tools by which the intricacy of the immune response can be explored. B cell epitopes are regions of the surface of a protein, or other biomacromolecule, recognized by soluble or membrane-bound Antibody molecules. In developing AntiJen, we have discarded the contents of our previous B cell archive and constructed one de novo. It contains an entirely new data set with a substantially different data structure. There are two forms of epitopes: linear and discontinuous. A linear B cell epitope is composed of a single stretch of sequential residues. A discontinuous B cell epitope is composed of sequentially separate residues brought into close proximity in a conformationally-dependent arrangement. The data we archive is primarily focused upon linear epitopes. This is due to the far greater amount of experimental data available for linear epitopes, which reflects both the relatively facile experiments needed to identify them and an implicit belief in their utility as potential vaccine epitopes. By contrast, discontinuous epitopes are thought to be more prevalent within folded proteins, but are far more challenging to determine experimentally. The archive catalogues the sequence of binding peptides, and also gives the length and source: TTGDVIASS, a 9 amino acid peptide from Escherichia coli non-fimbrial adhesion. Residues identified as important in binding to the antibody are recorded. This may correspond to a whole peptide or a subsequence, such as TTGDVI in the above example. The peptides are also categorized in terms of their relative observed immunodominance. Antibodies host organism and isotype are recorded. The current B cell epitope archive contains 3,541 epitopes.
T Cell Epitopes
T cell epitopes are short peptides bound by major histocompatibility complexes (MHC) and subsequently recognized by T cells. Epitopes recognized by both CD4+ and CD8+ T cells are included in the database. Such epitopes can be identified in many different ways. However, this diversity of measurement imposes a certain need for consistency, necessitating the requirement for recording a range of different experimental methods. The archive has expanded to include 4,158 entries. The entries contain the epitopes, ranging in length from 4 to 38 amino acids, peptide information, detailing the source, with links to Swiss-Prot and the corresponding MHC restriction data such as Serotype, Allele and Class. Additionally, the peptides are categorized in to groups such as Allergens, Bacterial, Cancer, Human, Viral and Self peptides.
MHC – Peptide binding
AntiJen continues to archive quantitative data on the thermodynamics of peptide interactions [18, 19], and it has expanded in number and content, with additions such as experimental conditions, plus specific Standard and Competitor peptide concentrations used in the assays. The current archive contains 15,454 entries. The sequence of the binding peptide, along with the source, plus relevant MHC restriction data is recorded. The restriction alleles currently include those from Human, Mouse, Rat, Rhesus Monkey, Cotton-top Tamarin, and Chimpanzee. AntiJen contains IC50 values, binding affinity measurements from competitive binding assays, for which the standard and competitor peptides and concentrations are recorded, plus BL50 values, calculated from peptide stabilizing assays. Where possible, antibodies and the concentrations used to calculate BL50 values are archived. Additionally, but on a somewhat smaller scale, equilibrium association (KA) and dissociation (KD) constants are recorded for peptide-MHC interaction. Melting temperatures (Tm) and signal wavelength are also recorded; this is the temperature and wavelength at which 50% of the MHC protein is denatured as measured by circular dichroism. AntiJen also records so-called Weak/Non-binders. This indicates that the peptide has been tested in an MHC restriction assay and has been found to exhibit a binding affinity, i.e. an IC50 value > 10,000 nM for a radio-ligand assay, so low that it can be categorized as inactive.
pMHC-T Cell Receptor interaction
The TCR sub-database contains 782 entries, which records thermodynamic and kinetic binding data for the interaction of peptide-MHC (pMHC) complexes with TCRs. Different MHCs exhibit a distinct selectivity for certain peptide sequences. T cell receptors, in their turn, also exhibit different affinities for peptide-MHC complexes. The entries contain epitope, peptide source and MHC restriction data, as described above, plus TCR structure information, located at the MMBD database http://www.ncbi.nlm.nih.gov/Structure/index.shtml. Furthermore, any mutations are noted and a designated name for the TCR is archived. In addition, the peptides are recorded as either agonists or antagonists. The binding data is given as equilibrium constants (KD), EC50 values, rate of association (Kon), rate of dissociation (Koff), association constant (KA) and the half-life (t1/2) of the TCR-peptide interaction.
TAP Binding
This dataset contains binding data for the interactions between peptides and the TAP transporter, one of the principle steps in antigen presentation. As with the peptide-MHC database, the data is established from competitor binding experiments, based on labelled assays. Therefore, standard and competitor peptide sequences and their concentrations are recorded. The binding data is given as IC50 and KD values. The database currently contains 1,106 entries, with peptides from Human, Rat and Mouse sources. Based on IC50 values > 10,000 nM, the peptides are categorized as weak/non-binders. The entries have increase in number from the level found in JenPep, although several entries were removed in an effort to increase the accuracy and consistency of the archive (Table 2).
Peptide-MHC Kinetics
AntiJen's kinetics sub-database, which contains 1,150 entries, mostly relates to Class I MHC data. It records measurements for forward and reverse rate constants for complexation events. This complements the thermodynamic measurements on peptide-MHC binding described above. The data currently focuses upon both the half-lives of binding interactions, as well as association and dissociation rate constant values (Kon and Koff) for the recorded epitopes. Additionally, concentrations of the peptide, MHC and TAP are archived. The half-life for radioisotope labelled β2-microglobulin dissociation from an MHC class I complex, as measured at 37°C, is also archived. This is a kinetic measurement rather than a thermodynamic one, although it is often assumed that the greater the half-life the stronger the peptide-MHC complex. The half-life (t1/2) equals:
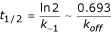
Here the t1/2 corresponds to the dissociation of the MHC-β2 microglobulin complex rather than the kinetics of the protein-ligand interaction, but is still peptide dependent, as well as kinetic in nature.
Immunological Protein-Protein Interactions
The immune system is built around protein interactions therefore we developed another new sub-database which deals with Immunological Protein-Protein Interactions (IPPI). This archive contains 2,675 entries based on a variety of binding data, such as Kon and Koff rates, for a range of macromolecules implicated in physiological or pathological interactions, as well as KD, KA and IC50 values. The molecules include receptors such as CD4 or CD8 molecules, superantigens and other microbial virulence factors, cytokine receptors and cell adhesion molecules. The entries detail both protein partners involved in the binding interaction, with links provided to the NCBI-Entrez database. Additional data for MHC receptors is archived, whereby the reactive epitope is recorded and the co-receptors are categorized into viral, bacterial and self peptides. MHC data outlined in the previous databases is given, where appropriate including any mutations to the MHC.
Antibody – Protein Binding
AntiJen also contains a further sub-database, which comprises thermodynamic data relating to antibody-antigen binding. The dataset contains 395 entries for antigen proteins and antibodies, mostly derived from viral and mammalian sources. Reported values were obtained using radiolabelled assays and BIAcore analysis. This archive should aid in the selection of antibodies and peptides for in vitro studies. The entries list the antibodies and the binding/kinetic data, consisting of KD and KA values and to a lesser extent Kon, Koff and IC50 values.
Peptide Libraries
This archive further complements our MHC binding databases by indicating the relative contribution of residues within peptide libraries to MHC binding. 897 entries contain quantitative specificity data derived from position specific peptide libraries [20]. This catalogues the relative effect on affinity, in the form of IC50, log relative SD50 and log SI values, of all substitutions, at all peptides positions, against a random sequence backdrop. All of the libraries relating to a known peptide binder are designated a name within AntiJen, this usually consists of the author and year of publication. The archive contains the core peptide, along with the mutation position and the substituted amino acid. The corresponding MHC data is given as mentioned above.
Diffusion Co-efficients
To further increase the range of data archived, AntiJen also contains 759 records of cellular biophysical data, in the form of diffusion co-efficients, recorded as cm2s-1, for a diversity of cell surface molecules, including MHCs (Mouse and Human), viral peptides and other receptors [21]. The molecules are either chemically or fluorescently labelled and then measured using one of two methods: Single Particle Tracking (SPT) or Fluorescence Photobleaching Recovery (FPR). SPT monitors the lateral motion of a labelled molecule while FPR measures the rate of subsequent infiltration from a photobleached section of the membrane. Friction co-efficient data is also given, which measures of the velocity and force applied to an antibody-coated bead. Records contain the cell or cell type where diffusion is occurring, the name of the diffusing protein along with the form of labelling applied. Furthermore, specific experimental data is given such as antibody bead size. In this case the diffusion of the beads is monitored. Increasingly, data relating to photobleaching is included, such as beam power, bleaching duration, pre- and post-bleach time, etc..
Copy Numbers
The final sub-database contains 414 measures of cell surface populations of different molecules, called cell surface copy numbers hereafter. This database focuses on an array of molecules, including Class I MHCs (22) and TCRs [23]. The entries are given as number and type of MHC molecules, number of MHC-peptide complexes or abundance of peptides associated with each MHC serotype, generally defined by mass spectroscopy. The entries list the cell type, the antibody bound to the MHC and, if appropriate, the binding epitope. This only applies to the number of MHC-peptide complexes and the abundance of peptides associated with each MHC.
Searching the database
Search mechanisms within AntiJen are significantly improved and allow either a detailed or a broad search from a simple user interface. From our experience with JenPep, we recognize that accessibility to the data in a user friendly manner is a vital requirement, and have improved our current search mechanisms and developed new search interfaces. Two different search mechanisms are available. One is based on BLAST [24] and the other is a bespoke system, allowing several alternative searches. Within a typical search, the user-entered search criteria are carried from an HTML form to a category specific PERL/SQL script, which performs the database queries.
The BLAST search allows querying of a peptide or nucleotide sequence against the proteins contained in AntiJen; all entries containing data within AntiJen which are relevant to a protein sequence are linked via the BLAST output. A local database of protein sequences is searched with BLASTP or BLASTX [24] using the BLOSUM64 matrix. All BLAST control variables are fixed. An HTML Front-End (Figure 2), where a sequence can be entered or uploaded, connects to a web server-based PERL/CGI scripts, which interacts with BLAST. An annotated version of the default BLAST output is produced and links to AntiJen entries via SWISS-PROT [12] accession codes, which act as a query within a Keyword search. This allows AntiJen entries to be viewed directly from the BLAST output.

Overview of the different search methods within AntiJen. The example search is focused upon an MHC ligand. The MHC ligand data can be searched directly (A) from a link on the AntiJen homepage, a broad search and specific search is available. A search for the epitope AMALLRLPLV, has one hit (D), this leads to the entry (G). All of the other sub-databases can be searched in this manner from the homepage. The two other searches are more generalised. A Keyword search (B) carries out a broad search on the whole database, for the criteria – Bacteria. This search gives 139 hits (E) and all of the sub-database entries can be selected from this output. The final search method is a BLAST search (C). The peptide (or nucleotide) sequence is queried against a local protein database. The output (F) provides links to the sub-databases relative to the protein.
At present, peptide string, keyword, and protein name index searches are available within the bespoke system, which allows querying of individual peptide sequences or at the level of whole protein antigens. An overview of the AntiJen search systems is given in Figure 2 and 3. Epitopes, MHC, TCR, TAP peptide binding and kinetics databases can all be searched using sequence strings. The search protocol first returns an epitope list and a count of epitope matches. Subsequently, experimental criteria can be accessed for each selected epitope. Peptides can be searched using an amino acid orientated query: a sparse peptide string, similar in form to a peptide binding motif [3] or a PROSITE pattern [25], is used to identify all matching sequences. See Figure 4. Alternately, a list of protein antigens within AntiJen can be searched using keywords; the thermodynamic binding data such as MHC-peptide, TCR-pMHC and TAP, plus the B and T cell archives, related to the search criteria can then be selected from the matches based upon the SWISS-PROT accession codes [12], displaying all corresponding entries in the database.The other search method, allows the IPPI, diffusion co-efficients, peptide libraries, antibody-protein and copy number sub-databases to be searched using an index method, within a user-friendly HTML drop-down menu. Each of these methods can also be moderated using subsidiary search filters, data size ranges, and result presentation alternatives, such as peptide length or IC50 values. Minimum and maximum values can be used to restrict results, as can selection of MHC restriction alleles.

Searchable database types within AntiJen. The database contains 3 types of searchable sub database: a set of Antigens searchable by keyword, various databases of functional and thermodynamic data searchable by peptide sequence, and a database of immunological protein-protein interactions searchable through an index. Peptide sequence searches can be explicit or "motif" based. Searches can also be focussed by setting the value ranges for properties, such as IC50 etc, recorded in the databases. Currently there is a link between all the thermodynamic and kinetic databases and between them and the database of antigens. Only antigens with data in one of the other sub-databases are included in the antigen database. Links to external databases are also indicated. The BLAST search provides an overall search of the databases except the Protein-Protein Interactions archive.

Sparse peptide sequence search. Examples of the two search types available in AntiJen: (Search 1) a substring query and (Searches 2 and 3) PROSITE-like sparse queries allowing sets of variable (indicated by asterisks) and alternative (encased in square brackets) amino acids in the search. In our example, each query is an extension of the previous search. The initial search type returns a single hit, which is a weak binder. By introducing variable amino acid positions within the query string, the second query permits access to a larger data set with 39 hits being returned. The third search utilises both variable and alternative amino acid request functionality and returns 1996 hits. The number of entries returned can be reduced by specifying the epitope length, limiting IC50 values and restricting by one MHC allele. This search is constrained to peptides of amino acid length 9, which returns 280 hits. Constraining further by MHC allele HLA-A*0201, reduces this to 120. An additional constraint using the IC50 data filter, and requesting values below 500 only (the epitope range), reduces this again to 60.
Discussion
The name given to our new database, AntiJen, reflects a shift from a peptide orientated database structure, which was inherent within our earlier JenPep database, to one which can properly balance its focus on both protein antigens and isolated peptides. As such, it represents an important, integrated, immunological data resource. AntiJen now provides broad insight into both T cell and B cell mediated antigen recognition. In addition, through the auspices of the IPPI sub-database, the database also throws light on co-stimulation by co-receptors and gives important insights into the innate immune response. Our approach to protein-protein interactions, focusing on measured affinities, complements other methods, such as the Yeast-2-Hybrid system, which, while giving greater volumes of data, has problems of accuracy [26]. This, while not devoid of experimental artifact, gives a usefully different perspective on cataloguing protein-protein interactions. The further addition of weak binding notation to the MHC-peptide and TAP provides a greater overview of the nature of antigenic epitopes. This is further improved by the addition of the peptide libraries database, whereby key peptide residues can be highlighted. New databases have expanded the breadth of AntiJen to include biophysical data such as diffusion co-efficients and cellular data such as abundance of molecules. The antibody-antigenic protein sub-database will also provide a key resource for in vivo and in vitro studies, aiding in the selection of antibodies and peptide/protein targets.
AntiJen distinguishes itself from the other specific binding databases [2–8] in several ways. Firstly, more data is recorded; our MHC-peptide database contains over 2,000 more entries than MHCPEP [2] and 10,000 more entries than EPIMHC [7]. Additionally, we have not restricted our archive to only high binders or to a specific category, as seen in EPIMHC and the HIV sequence databases [5]. Furthermore, AntiJen is currently a curated database, which is constantly expanding.
Most obviously, AntiJen is useful in the design of epitope and subunit vaccines. Additionally, AntiJen is helpful in the design of clinical diagnostics and other laboratory reagents, such as the selection of peptides for tetramer design. AntiJen is also useful in the parameterization of mathematical models in theoretical immunology [27]. The redevelopment of the database has focused not only on content, but also on infrastructure. The current system, based on epitope string, keyword and index searches, along with an overall BLAST search, plus the redesigned HTML interface, leads to much greater accessibility and usability. Finally, the database acts as a repository of quantitative, continuous data, for the development of data-driven in silico predictive models, such as prediction of epitopes and MHC binding [18, 19, 28, 29] through QSAR modeling.
Future work
Future tasks in the development of AntiJen, fall into two principle categories: eliminating deficiencies, errors, and inconsistencies within the database and simultaneously reinforcing it by expanding its depth, breadth, and scope. We also need to monitor updates within external databases, so that any alterations are mirrored within the archive. Like all other such repositories, AntiJen is prone to both systematic and random errors within the data accumulation process. User feedback and our interactions with immunologists will hopefully address persisting errors. Deficiencies in our database include our current inability to encode chemically or post-translationally modified peptides, non-natural MHC mutants and non-amino acid peptidomimetic MHC ligands. Additionally, it would also be interesting to complement our existing data on TAP binding with information on antigen presentation pathways, such as proteasomal and cathepsin cleavage patterns. Moreover, the compilation of B cell or antibody epitope data is an area ripe for robust development. Linear and conformational B cell epitopes are very much larger in number than our current compilation, leaving us scope to greatly increase recorded epitopes.
Conclusion
The development of a database is always a work in progress. Not simply because the easily accessible literature is typically always increasing, but also because of the desire to capture as much of the existing, but hidden, literature, as possible. In the post-genomic era, the database has formed the bedrock and language of bioinformatics; increasingly databases are coming to underpin our modern understanding of biology as a whole. Traditionally, databases have arisen as a response to need, answering the individual and idiosyncratic questions posed by biologists. However, the history of bioinformatics databases has shown the extraordinarily diverse ways in which archived data can be used.
In creating AntiJen, we were motivated partly by our desire, and the desire of collaborators, to use the data within it to build predictive in silico models [16, 17, 28, 29], and partly by a more altruistic desire to generate a useful, integrated database system with a quantitative focus. AntiJen has many potential uses throughout the immunological discipline, from immunoinformaticans to experimental immunologists and vaccinologists. By increasing the degree to which data is machine readable and web accessible, we open up new, and previously unthought-of, avenues for the bioinformatic exploration of immunological data.
AntiJen is a primary data resource, amongst the most complete of its kind, yet, like SWISSPROT [12] or GenBank [30] decades ago, it is still relatively small and offers much scope for improved annotation. We see the database as a foundation from which to consolidate, through time, thus achieving a comprehensive resource of immunological data.
References
Brusic V, Zeleznikow J, Petrovsky N: Molecular immunology databases and data repositories. J Immunol Methods 2000, 238:17–28.
Brusic V, Rudy G, Harrison LC: MHCPEP, a database of MHC-binding peptides: update 1997. Nucleic Acids Res 1998, 26:368–371.
Schonbach C, Koh JL, Flower DR, Wong L, Brusic V: FIMM, a database of functional molecular immunology: update 2002. Nucleic Acids Res 2002, 30:226–229.
Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S: SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 1999, 50:213–219.
Kuiken C, Korber B, Shafer RW: HIV sequence databases. AIDS Rev 2003, 5:52–61.
Sathiamurthy M, Hickman HD, Cavett JW, Zahoor A, Prilliman K, Metcalf S, Fernandez Vina M, Hildebrand WH: Population of the HLA ligand database. Tissue Antigens 2003, 61:12–19.
Reche PA, Zhang H, Glutting JP, Reinherz EL: EPIMHC: a curated database of MHC-binding peptides for customized computational vaccinology. Bioinformatics 2005, 21:2140–2141.
Bhasin M, Singh H, Raghava GPS: MHCBN: a comprehensive database of MHC binding and non-binding peptides. 2002.
Blythe MJ, Doytchinova IA, Flower DR: JenPep: a database of quantitative functional peptide data for immunology. Bioinformatics 2002, 18:434–439.
McSparron H, Blythe MJ, Zygouri C, Doytchinova IA, Flower DR: JenPep: A Novel Computational Information Resource for Immunobiology and Vaccinology. J Chem Inf Comput Sci 2003, 43:1276–1287.
Blythe MJ, Flower DR: Benchmarking B cell epitope prediction: underperformance of existing methods. Protein Sci 2005, 14:246–248.
Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O'Donovan C, Phan I, Pilbout S, Schneider M: The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res 2003, 31:365–370.
Govindarajan KR, Kangueane P, Tan TW, Ranganathan S: MPID: MHC-Peptide Interaction Database for sequence-structure-function information on peptides binding to MHC molecules. Bioinformatics 2003, 19:309–310.
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE: The Protein Data Bank. Nucleic Acids Res 2000, 28:235–242.
Robinson J, Waller MJ, Parham P, de Groot N, Bontrop R, Kennedy LJ, Stoehr P, Marsh SG: IMGT/HLA and IMGT/MHC: sequence databases for the study of the major histocompatibility complex. Nucleic Acids Res 2003, 31:311–314.
Gilks WR, Audit B, De Angelis D, Tsoka S, Ouzounis CA: Modeling the percolation of annotation errors in a database of protein sequences. Bioinformatics 2002, 18:1641–1649.
Linial M: How incorrect annotations evolve – the case of short ORFs. Trends Biotechnol 2002, 21:298–300.
Doytchinova IA, Flower DR: A comparative molecular similarity index analysis (CoMSIA) study identifies an HLA-A2 binding supermotif. J Comput Aided Mol Des 2002, 16:535–544.
Doytchinova IA, Flower DR: Towards the quantitative prediction of T-cell epitopes: coMFA and coMSIA studies of peptides with affinity for the class I MHC molecule HLA-A*0201. J Med Chem 2001, 44:3572–3581.
Liu R, Enstrom AM, Lam KS: Combinatorial peptide library methods for immunobiology research. Exp Hematol 2003, 31:11–30.
Chakrabarti A, Matko J, Rahman NA, Barisas BG, Edidin M: Self-association of class I major histocompatibility complex molecules in liposome and cell surface membranes. Biochemistry 1992, 31:7182–7189.
Lippolis JD, White FM, Marto JA, Luckey CJ, Bullock TN, Shabanowitz J, Hunt DF, Engelhard VH: Analysis of MHC class II antigen processing by quantitation of peptides that constitute nested sets. J Immunol 2002, 169:5089–5097.
Marano N, Holowka D, Baird B: Bivalent binding of an anti-CD3 antibody to Jurkat cells induces association of the T cell receptor complex with the cytoskeleton. J Immunol 1989, 143:931–938.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997, 25:3389–3402.
Gattiker A, Gasteiger E, Bairoch A: ScanProsite: a reference implementation of a PROSITE scanning tool. Appl Bioinformatics 2002, 1:107–108.
Sprinzak E, Sattath S, Margalit H: How reliable are experimental protein-protein interaction data? J Mol Biol 2003, 327:919–923.
Andersen PS, Menne C, Mariuzza RA, Geisler C, Karjalainen K: A response calculus for immobilized T cell receptor ligands. J Biol Chem 2001, 276:49125–49132.
Guan P, Doytchinova IA, Flower DR: HLA-A3 supermotif defined by quantitative structure-activity relationship analysis. Protein Eng 2003, 16:11–18.
Doytchinova IA, Blythe MJ, Flower DR: Additive method for the prediction of protein-peptide binding affinity. Application to the MHC class I molecule HLA-A*0201. J Proteome Res 2002, 1:263–272.
Benson DA, Karsch-Mizradu I, Lipman DJ, Ostell J, Rapp BA, Wheeler DL: GenBank. Nucleic Acid Res 2000, 28:15–18.
Acknowledgements
We acknowledge the helpful assistance of Ms Christianna Zygouri. We should also like to thank Dr P Borrow and Prof V Brusic for helpful discussions. The Edward Jenner Institute for Vaccine Research wishes to thank its sponsors: GlaxoSmithKline, the Medical Research Council, the Biotechnology and Biological Sciences Research Council, and the UK Department of Health.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Toseland, C.P., Clayton, D.J., McSparron, H. et al. AntiJen: a quantitative immunology database integrating functional, thermodynamic, kinetic, biophysical, and cellular data. Immunome Res 1, 4 (2005). https://doi.org/10.1186/1745-7580-1-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1745-7580-1-4